[本站讯]近日,齐鲁医院神经内科焉传祝/纪坤乾教授团队在神经系统罕见病研究领域取得了系列新进展,研究成果先后发表于自然科学综合类顶刊Science Bulletin(中科院一区,IF=21.1)、临床神经病学领域顶级期刊JAMA Neurology(中科院一区,5年IF=22.5),神经病学领域顶级期刊Neurology(中科院一区,5年IF=9.3)。
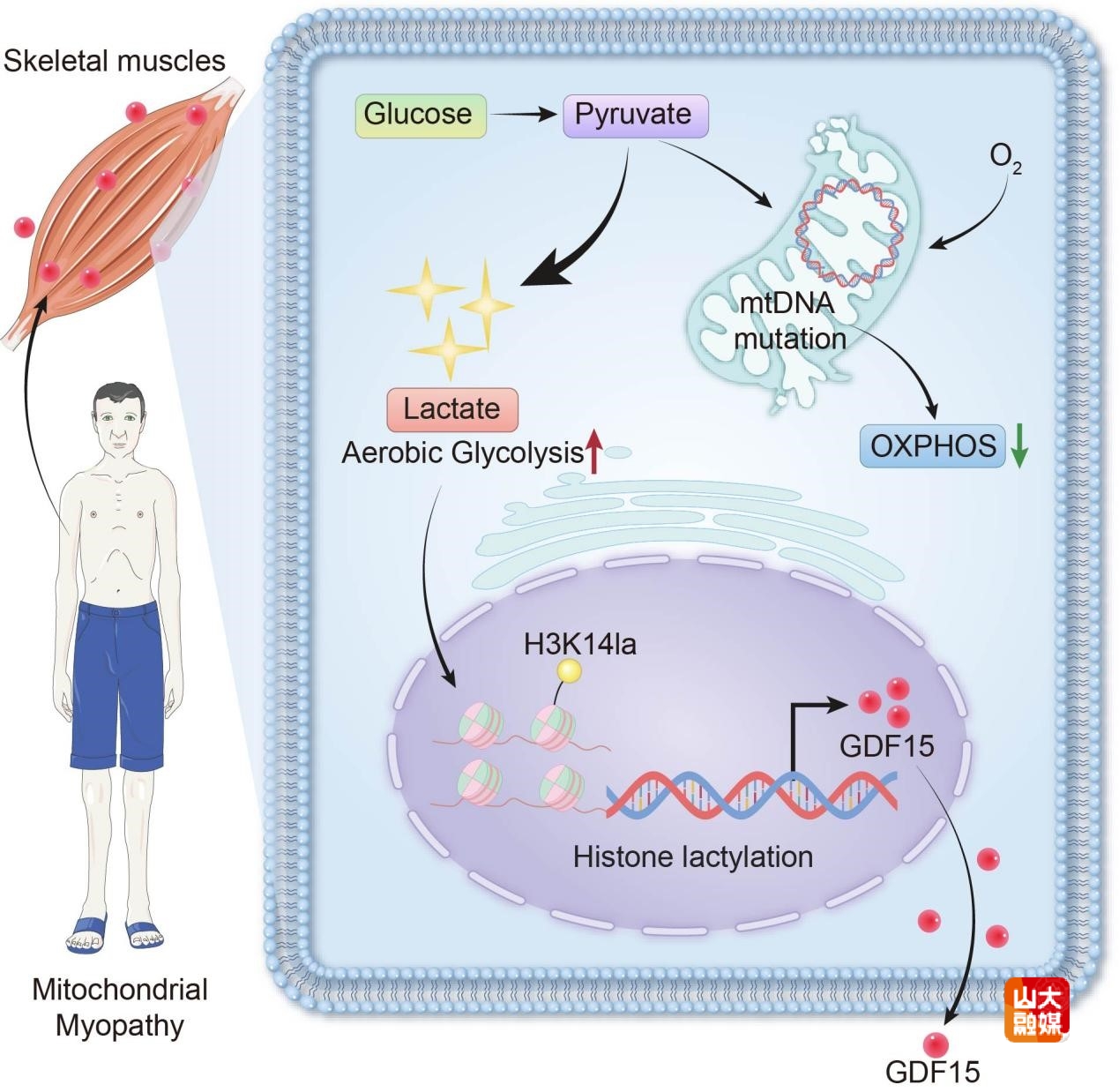
线粒体DNA(mtDNA)突变是导致线粒体病(mitochondrial disease, MD),尤其是线粒体肌病(mitochondrial myopathy, MM)发病的核心分子基础。mtDNA突变可以导致氧化磷酸化功能受损,引起能量代谢障碍,同时伴随乳酸的病理性过量积累。近年来,乳酸已被发现不仅是代谢副产物,更可作为信号分子参与表观遗传调控。与此同时,生长分化因子15(Growth Differentiation Factor 15,GDF15)作为线粒体应激的关键细胞因子,在mtDNA突变相关疾病中持续性表达升高。然而,乳酸如何通过表观遗传途径精确调控GDF15的表达与分泌,这一关键分子机制此前尚未得到系统解析。团队首次阐明在mtDNA突变引发的线粒体应激条件下,乳酸通过驱动组蛋白H3K14乳酸化修饰(H3K14la)调控骨骼肌GDF15分泌的分子机制,为深入解析MM的发病机制、挖掘潜在治疗干预靶点提供了全新的理论支撑与实验依据。齐鲁医院神经内科林岩、李步苏为共同第一作者;焉传祝教授、纪坤乾副主任医师,中国科学院广州生物医药与健康研究院教授刘兴国为该文共同通讯作者;山东大学齐鲁医院为该文的第一和通讯作者单位。
团队首次报道视神经脊髓炎(NMOSD)患者 AQP4-IgG 检测中,因前带效应(Prozone Effect)出现未稀释及 1:10 稀释时假阴性,1:100 及 1:5000 稀释后呈阳性的罕见现象,还发现患者存在视神经炎症状与脑内弥漫白质、基底节区病灶的非典型影像学矛盾表现。通过体外实验证实前带效应可致 AQP4-IgG 假阴性,且用其他患者纯化 AQP4-IgG 验证了该效应。该研究报道有重要临床应用价值,提示临床检测 AQP4-IgG 需行系列稀释,避免漏诊;对疑似 NMOSD 但 AQP4-IgG 阴性者,需考虑前带效应并进一步检测,同时拓展 NMOSD 影像学认知,助力与其他神经系统疾病鉴别,指导临床诊疗。齐鲁医院神经内科主治医师娄建伟、教授曹丽丽为共同第一作者,纪坤乾为论文通讯作者。山东大学齐鲁医院为该文的第一和通讯作者单位。
团队报道了一位45岁男性患者,以肢体麻木、头晕就诊,详细的神经系统体格检查发现眼球扑动(ocular flutter)的体征,初步血清学、脑 MRI 等检查无异常,但脑脊液出现蛋白细胞分离现象。团队通过针对性自身抗体检测,确诊其为抗 GD1b 和抗 GT1a 抗体阳性的脑干脑炎(Bickerstaff encephalitis)。经静脉注射免疫球蛋白治疗,患者症状显著改善。该案例首次报道了抗 GD1b/GT1a 抗体相关脑干脑炎患者眼球扑动的体征,为类似疑难罕见病例提供精准诊断思路。齐鲁医院神经内科进修医师秦瑶、硕士研究生徐志鸿为共同第一作者,纪坤乾副主任医师为论文通讯作者。山东大学齐鲁医院为该文的第一和通讯作者单位。
山东大学齐鲁医院神经内科焉传祝教授引领的神经系统罕见病团队长期致力于神经系统罕见病的临床与基础研究,坚持以临床问题为导向,提出科学问题并进行相关的基础及临床研究。对包括脂质沉积性肌病、线粒体病、糖原累积病、炎症性肌病、肌营养不良、钴胺素代谢缺陷、晚发型脑白质营养不良及肌萎缩侧索硬化等多种神经系统罕见病的临床特点、病理生理机制及治疗靶点方面进行了系统、深入的探索。

















